

Researchers have discovered how a cell surface protein called Aplp1 can play a role in spreading material responsible for Parkinson’s disease from cell-to-cell in the brain.
Promisingly, an FDA-approved cancer drug that targets another protein called Lag3 – which interacts with Aplp1 – blocks the spread in mice, suggesting a potential therapy may already exist.
In a recent paper, an international team of scientists describes how the two proteins work together to help harmful alpha-synuclein protein clumps get into brain cells.
“Now that we know how Aplp1 and Lag3 interact, we have a new way of understanding how alpha-synuclein contributes to the disease progression of Parkinson’s disease,” neuroscientist Xiaobo Mao from Johns Hopkins University said in June.
“Our findings also suggest that targeting this interaction with drugs could significantly slow the progression of Parkinson’s disease and other neurodegenerative diseases.”
More than 8.5 million people globally have Parkinson’s, the second most common neurodegenerative disease after Alzheimer’s.
As a progressive movement disorder, it’s usually only diagnosed when symptoms show, which include tremors, stiffness, balance problems, speech difficulties, disturbed sleep patterns, and mental health issues. Currently incurable, the disease means patients may eventually struggle to walk or speak.
Parkinson’s symptoms mainly result from the death or impairment of dopamine-producing neurons in the brain’s substantia nigra, a region involved in fine motor control. This is thought to be caused by Lewy bodies, which are abnormal clumps of protein mostly consisting of misfolded alpha-synuclein that travel between neurons.
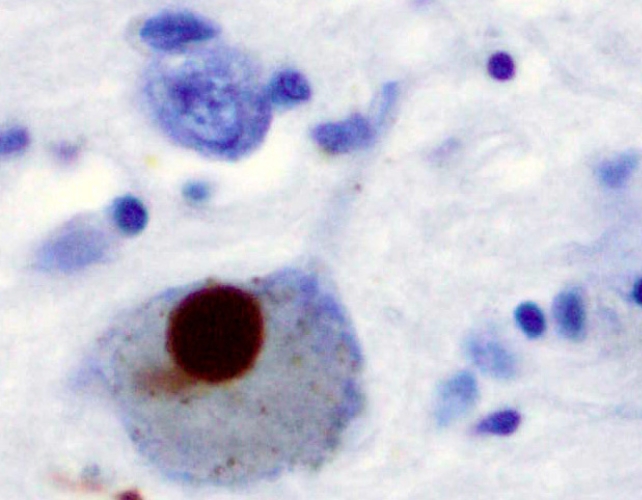
Alpha-synuclein typically maintains functional communication between neurons, but problems arise when it becomes misfolded and insoluble. That said, identifying whether this is a cause of Parkinson’s or a symptom is difficult.
Past studies on mice found Lag3 binds to alpha-synuclein proteins and spreads Parkinson’s disease pathology in neurons. While deleting Lag3 significantly impedes this process, it does not completely prevent it, indicating another protein was also implicated in neurons taking in misfolded alpha-synuclein.
“Our work previously demonstrated that Lag3 wasn’t the only cell surface protein that helped neurons absorb alpha-synuclein, so we turned to Aplp1 in our most recent experiments,” said Johns Hopkins neuroscientist Valina Dawson.
The scientists conducted tests with genetically modified mice that were missing either Aplp1 or Lag3, or both. They found Aplp1 and Lag3 can each independently help brain cells absorb harmful alpha-synuclein, but together they significantly increase the uptake.
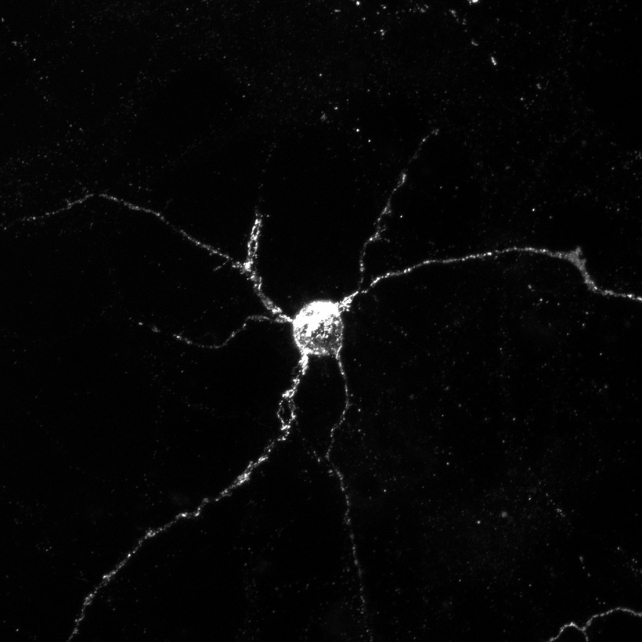
When mice were missing both Aplp1 and Lag3, 90 percent less of the harmful alpha-synuclein entered healthy brain cells, meaning a greater amount of the harmful protein clumps was blocked with both proteins missing compared with a deletion of just one.
The researchers gave normal mice the drug nivolumab/relatlimab, a melanoma medication that contains a Lag3 antibody, and found that it also stopped Aplp1 and Lag3 from interacting, again almost completely blocking the formation of disease-causing alpha-synuclein clumps in neurons.
“The anti-Lag3 antibody was successful in preventing further spread of alpha-synuclein seeds in the mouse models and exhibited better efficacy than Lag3-depletion because of Aplp1’s close association with Lag3,” said Ted Dawson, a neuroscientist at Johns Hopkins University.
The next step will be to test the Lag3 antibody on mouse models of Parkinson’s disease and Alzheimer’s – where research has pointed to Lag3 as a target too.


